



PCR (polymerase chain reaction) is one of the in-vitro DNA amplification technologies, with a history of more than 30 years.
PCR technology was pioneered by Kary Mullis of Cetus, USA in 1983. Mullis applied for a PCR patent in 1985 and published the first PCR academic paper on Science in the same year. Mullis was awarded the Nobel Prize in chemistry in 1993 for his work.
Basic Principles of PCR
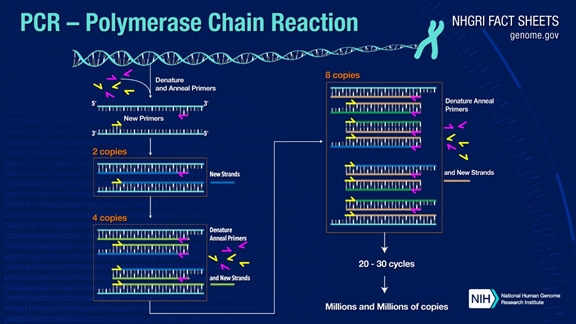
PCR can amplify target DNA fragments by more than one million times. The principle is under the catalysis of DNA polymerase, using parent strand DNA as a template and specific primer as the starting point for extension. It is replicated in vitro through steps such as denaturation, annealing, and extension. The process of daughter strand DNA complementary to the parent strand template DNA.

The standard PCR process is divided into three steps:
1.Denaturation: Use high temperature to separate DNA double strands. The hydrogen bond between DNA double strands is broken at high temperature (93-98℃).
2.Annealing: After the double-stranded DNA is separated, lower the temperature so that the primer can bind to the single-stranded DNA.
3.Extension: The DNA polymerase starts to synthesize complementary strands along the DNA strands from the primers bound when the temperature is lowered. When the extension is completed, a cycle is completed, and the number of DNA fragments doubles
Reciprocating these three steps 25-35 times, the number of DNA fragments will increase exponentially.
The ingenuity of PCR is that different primers can be designed for different target genes, so that target gene fragments can be amplified in a short period of time.
So far, PCR can be divided into three categories, namely ordinary PCR, fluorescent quantitative PCR and digital PCR.
The first generation of ordinary PCR
Use an ordinary PCR amplification instrument to amplify the target gene, and then use agarose gel electrophoresis to detect the product, only qualitative analysis can be done.
The main disadvantages of the first generation PCR:
1.Prone to non-specific amplification and false positive results.
2.The detection takes a long time and the operation is cumbersome.
3.Only qualitative test can be done
Second-generation Real-Time PCR
Real-Time PCR, also known as qPCR, uses fluorescent probes that can indicate the progress of the reaction system, and monitors the accumulation of amplified products through the accumulation of fluorescent signals, and judges the results through the fluorescence curve. It can be quantified with the help of Cq value and standard curve.
Because the qPCR technology is carried out in a closed system, the probability of contamination is reduced, and the fluorescence signal can be monitored for quantitative detection, so it is the most widely used in clinical practice and has become the dominant technology in PCR.
The fluorescent substances used in real-time fluorescent quantitative PCR can be divided into: TaqMan fluorescent probe, molecular beacons and fluorescent dye.
1)TaqMan fluorescent probe:
During PCR amplification, a specific fluorescent probe is added while adding a pair of primer. The probe is an oligonucleotide, and both ends are labeled with a reporter fluorescent group and a quencher fluorescent group.
When the probe is intact, the fluorescent signal emitted by the reporter group is absorbed by the quenching group; during PCR amplification, the 5′-3′ exonuclease activity of Taq enzyme cleaves and degrades the probe, making the reporter fluorescent group and quencher The fluorescent group is separated, so that the fluorescence monitoring system can receive the fluorescence signal, that is, every time a DNA strand is amplified, a fluorescent molecule is formed, and the accumulation of the fluorescence signal is completely synchronized with the formation of the PCR product.
2) SYBR fluorescent dye:
In the PCR reaction system, an excess of SYBR fluorescent dye is added. After the SYBR fluorescent dye is non-specifically incorporated into the DNA double-strand, it emits a fluorescent signal. The SYBR dye molecule that is not incorporated into the chain will not emit any fluorescent signal, thereby ensuring the fluorescent signal The increase in PCR products is completely synchronized with the increase in PCR products. SYBR only binds to double-stranded DNA, so the melting curve can be used to determine whether the PCR reaction is specific.

3) Molecular beacon:
It is a stem-loop double-labeled oligonucleotide probe that forms a hairpin structure of about 8 bases at the 5 and 3 ends. The nucleic acid sequences at both ends are complementarily paired, causing the fluorescent group and the quenching group to be tight. Close, no fluorescence will be produced.

After the PCR product is generated, during the annealing process, the middle part of the molecular beacon is paired with a specific DNA sequence, and the fluorescent gene is separated from the quencher gene to produce fluorescence.

The main disadvantages of second-generation PCR:
Sensitivity is still lacking, and the detection of low-copy specimens is inaccurate.
There is the influence of the background value, and the result is susceptible to interference.
When there are PCR inhibitors in the reaction system, the detection results are susceptible to interference.
Third generation digital PCR
Digital PCR (DigitalPCR, dPCR, Dig-PCR) calculates the copy number of the target sequence through end-point detection, and can perform accurate absolute quantitative detection without using internal controls and standard curves.
Digital PCR uses end-point detection and does not depend on the Ct value (cycle threshold), so the digital PCR reaction is less affected by the amplification efficiency, and the tolerance to PCR reaction inhibitors is improved, with high accuracy and reproducibility.
Due to the characteristics of high sensitivity and high accuracy, it is not easily interfered by PCR reaction inhibitors, and it can achieve true absolute quantification without standard products, which has become a research and application hotspot.
According to the different forms of the reaction unit, it can be divided into three main types: microfluidic, chip and droplet systems.
1) Microfluidic digital PCR, mdPCR:
Based on the microfluidic technology, the DNA template is separated. The microfluidic technology can realize the sample nano-upgrading or the generation of smaller droplets, but the droplets need a special adsorption method and then combined with the PCR reaction system. mdPCR has gradually been adopted by other methods replace.
2) Droplet-based digital PCR, ddPCR:
Use water-in-oil droplet generation technology to process the sample into droplets, and divide the reaction system containing nucleic acid molecules into thousands of nanoscale droplets, each of which does not contain the nucleic acid target molecule to be detected, or Contains one to several nucleic acid target molecules to be tested.
3) Chip-based digital PCR, cdPCR:
Use the integrated fluid pathway technology to engrave many microtubes and microcavities on silicon wafers or quartz glass, and control the flow of the solution through different control valves, and divide the sample liquid into nanometers of the same size into the reaction wells for digital PCR Reaction to achieve absolute quantification.
The main disadvantages of the third generation PCR:
The equipment and reagents are expensive.
The template quality requirements are high. If the template quantity exceeds the microsystem quantity, it will be impossible to quantify, and if it is too small, the quantification accuracy will be reduced.
False positives can also be generated when there is non-specific amplification.
Post time: Jul-30-2021



